*Based on cumulative number of patient-years of exposure observed across published Phase 2–4 clinical studies in relevant targeted immunomodulator (TIM) therapies2
WELL-STUDIED
SAFETY
PROFILE
INDICATIONS1
RINVOQ is indicated for the treatment of:
- Moderately to severely active rheumatoid arthritis (RA) in adults who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers.
- Active ankylosing spondylitis (AS) in adults who have had an inadequate response or intolerance to one or more TNF blockers.
- Active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation in adults who have had an inadequate response or intolerance to TNF blocker therapy.
- Giant cell arteritis (GCA) in adults.
Limitations of Use: RINVOQ is not recommended for use in combination with other Janus kinase (JAK) inhibitors, biologic disease-modifying antirheumatic drugs (bDMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine.
RINVOQ/RINVOQ LQ is indicated for the treatment of:
- Active psoriatic arthritis (PsA) in adults and pediatric patients 2 years of age and older who have had an inadequate response or intolerance to one or more TNF blockers.
- Active polyarticular juvenile idiopathic arthritis (pJIA) in patients 2 years of age and older who have had an inadequate response or intolerance to one or more TNF blockers.
Limitations of Use: RINVOQ is not recommended for use in combination with other Janus kinase (JAK) inhibitors, biologic disease-modifying antirheumatic drugs (bDMARDs), or with potent immunosuppressants such as azathioprine and cyclosporine.
Approved in 9 INDICATIONS
RA, PsA, AS, nr-axSpA, pJIA, GCA, AD, UC & CD1
12 YEARS
of clinical experience3*
>15,000 patients in 27 global clinical trials
across US-approved indications, including pediatrics 2+ years in pJIA and older patients (mean age 71) in GCA1,4,5
>47,000
patient-years of exposure to RINVOQ (15, 30, 45 mg) or RINVOQ LQ (1 mg/mL)4
~9.5 YEARS
maximum exposure in RA (~4.2 years median) to RINVOQ 15 mg as of August 15, 20254†
*Clinical experience encompasses the time from first RINVOQ patient dosed in RA clinical trial to present.
†In PsA: ~6.4 years maximum exposure (~3.6 years median) to RINVOQ 15 mg as of August 15, 2025. In AS: ~3.8 years maximum exposure (~1.8 years median) to RINVOQ 15 mg as of August 15, 2025. In nr-axSpA: ~2.3 years maximum exposure (~1.0 year median) to RINVOQ 15 mg as of August 15, 2025.4
AD=atopic dermatitis; AS=ankylosing spondylitis; CD=Crohn’s disease; GCA=giant cell arteritis; IR=intolerance or inadequate response; nr-axSpA=non-radiographic axial spondyloarthritis; pJIA=polyarticular juvenile idiopathic arthritis; PsA=psoriatic arthritis; RA=rheumatoid arthritis; TNFi=tumor necrosis factor inhibitor; UC=ulcerative colitis.
Please see Important Safety Information, including BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis, below.
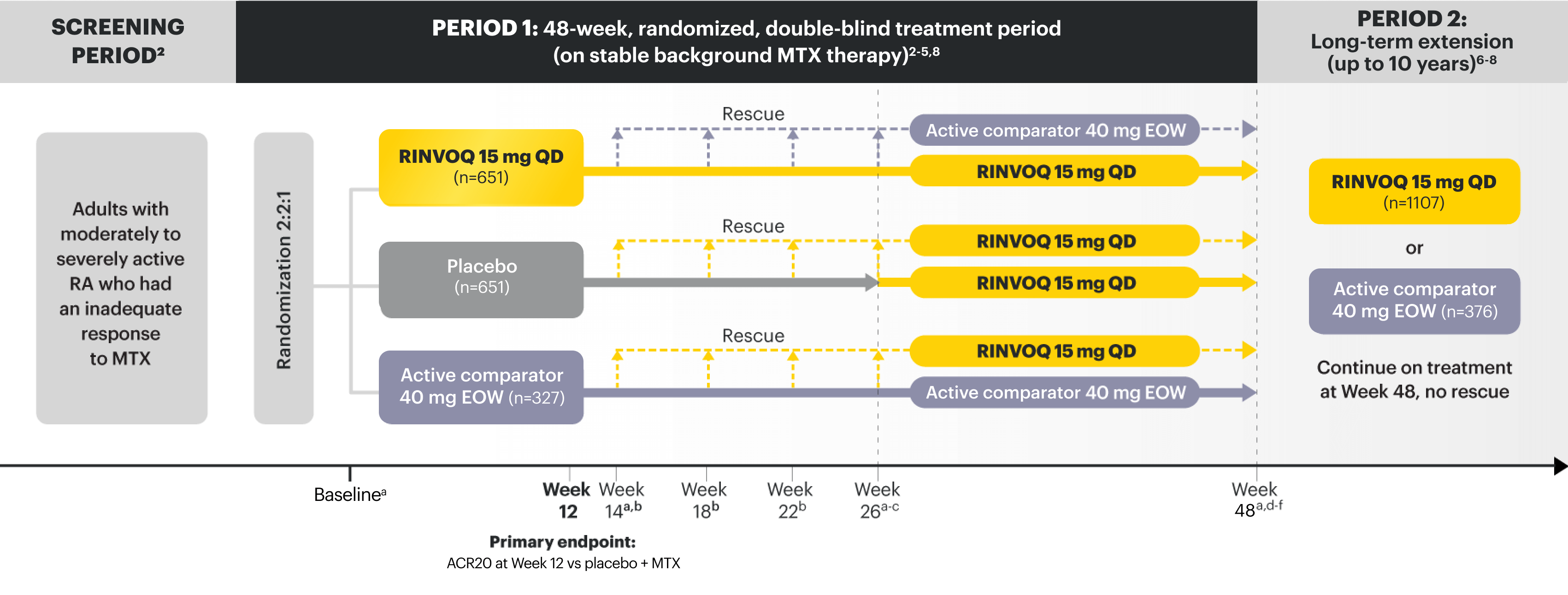
Well-Studied Safety Profile With Up to 12 Months of Data
Adverse Events of Special Interest at Weeks 12/14 and Week 52 in RA1,6
<<Swipe table to see more
| Week 12/14 | Week 12/14 | Week 52 | |||||
|---|---|---|---|---|---|---|---|
| Placebo-controlled trials NEXT, COMPARE, and BEYOND |
MTX-controlled trials EARLY and MONOTHERAPY |
Pooled 12-month data EARLY, MONOTHERAPY, NEXT, and BEYOND |
|||||
| TEAEs of Special Interest* n/100 PYsa (n/PYs) |
Placebo + csDMARDs n=1042 |
RINVOQ 15 mg QD + csDMARDs n=1035 |
MTX n=530 |
RINVOQ 15 mg QD n=534 |
RINVOQ 15 mg QD n=1213 |
||
| INFECTIONS | |||||||
| Serious infections | 2.3 (6/256.6) | 4.6 (12/258.3) | 1.6 (2/121.6) | 2.4 (3/126.4) | 3.5 (38/1081.6) | ||
| Active tuberculosis (TB) | 0 | 0 | 0 | 0 | 0.18 (2/1089.2) | ||
| Opportunistic infection (excluding TB) | 1.2 (3/256.8) | 1.9 (5/258.5) | 0.8 (1/121.5) | 0 | 0.6 (7/1088.9) | ||
| Herpes zoster | 1.2 (3/256.4) | 2.7 (7/258.7) | 1.6 (2/121.4) | 4.8 (6/126.0) | 3.8 (41/1073) | ||
| MALIGNANCY | |||||||
| Malignancy (excluding NMSC) | 0.4 (1/256.8) | 0.4 (1/259.3) | 0.8 (1/121.6) | 2.4 (3/126.8) | 1.2 (13/1090.2) | ||
| Lymphoma | 0 | 0 | 0 | 0.8 (1/126.8) | <0.1 (1/1091.6) | ||
| NMSC | 0.4 (1/256.7) | 0 | 0.8 (1/121.7) | 0 | 0.4 (4/1090.4) | ||
| CARDIOVASCULAR EVENTS | |||||||
| Adjudicated VTEb | 0.4 (1/256.8) | 0.8 (2/259.2) | 0 | 0.8 (1/126.7) | 0.5 (5/1090.8) | ||
| Adjudicated MACEc | 1.2 (3/256.8) | 0.4 (1/259.3) | 0.8 (1/121.7) | 1.6 (2/126.8) | 0.8 (9/1090.1) | ||
| GASTROENTEROLOGICAL EVENTS | |||||||
| Adjudicated GI perforations | 0 | 0 | 0 | 0 | <0.1 (1/1091.4) | ||
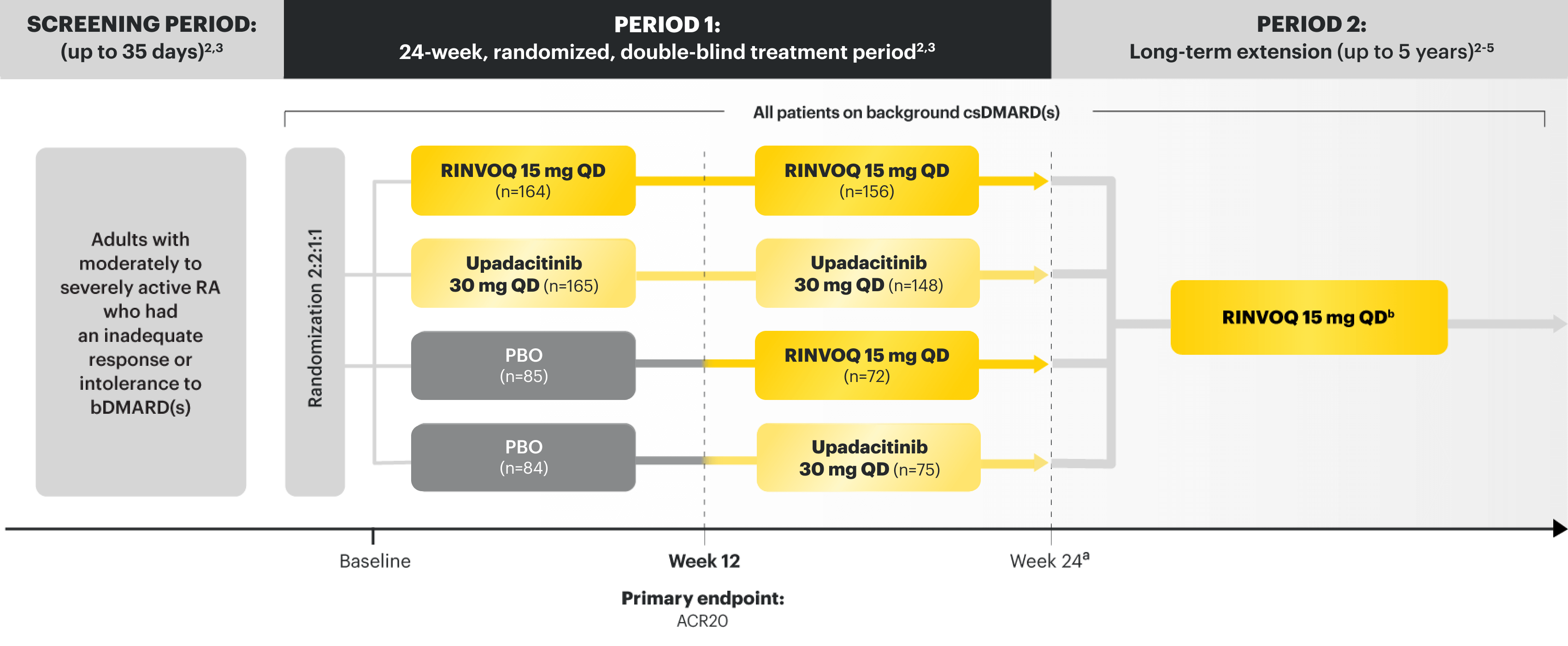
Data from 5 robust phase 3 clinical trials in RA1
Patients could advance or switch to RINVOQ from placebo, or be rescued to RINVOQ from active comparator or placebo as early as Week 12, depending on the study design.1
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
*TEAE=treatment-emergent adverse event is defined as any adverse event with an onset date on or after the first dose of study drug and no more than 30 days after the last dose of study drug if subject discontinued study drug prematurely.6
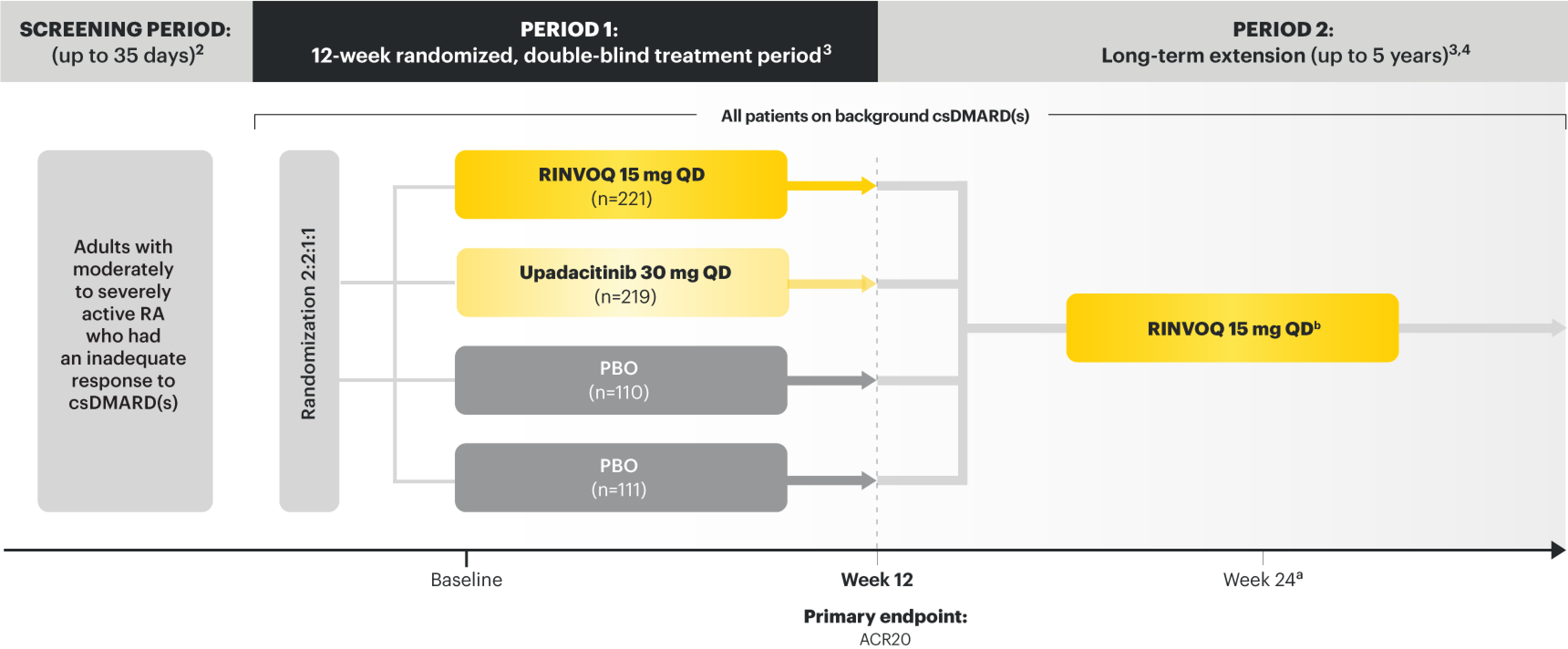
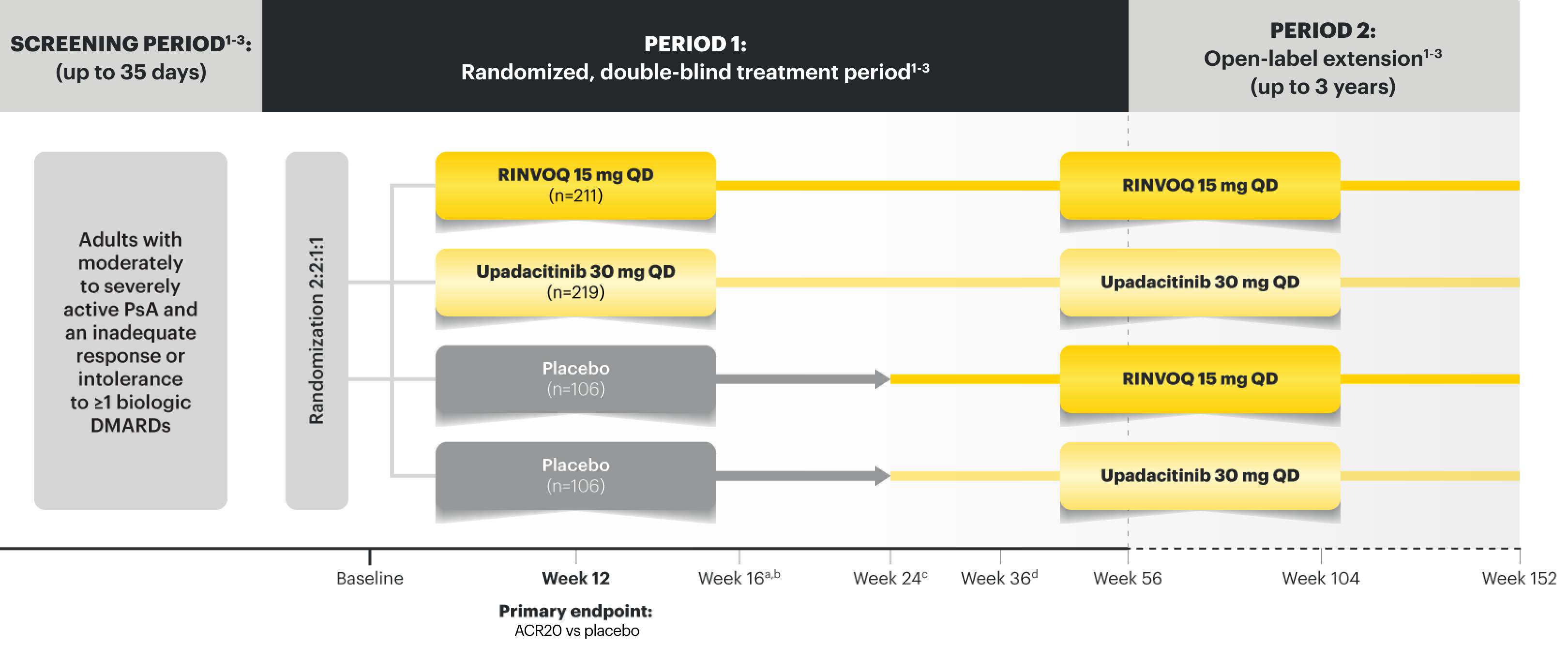
Adverse Events of Special Interest at Week 24 in PsA11*
<<Swipe table to see more
| SELECT-PsA 211 (bDMARD-IR) | SELECT-PsA 1 & SELECT-PsA 2 POOLED11 (non-bDMARD-IR & bDMARD-IR) | ||||||
|---|---|---|---|---|---|---|---|
| TEAEs of Special Interest† n/100 PYs (n/PYs) |
Placebo n=212 |
RINVOQ 15 mg QD n=211 |
Placebo n=635 |
RINVOQ 15 mg QD n=640 |
|||
| INFECTIONS | |||||||
| Serious infections | 1.2 (1/85.1) | 1.1 (1/91.9) | 1.9 (5/268.0) | 2.1 (6/281.2) | |||
| Active tuberculosis (TB) | 0 | 0 | 0 | 0 | |||
| Opportunistic infection (excluding TB and herpes zoster) |
0 | 0 | 0 | 0.4 (1/281.7) | |||
| Herpes zoster | 2.4 (2/84.8) | 3.3 (3/91.1) | 1.9 (5/267.9) | 2.5 (7/280.5) | |||
| MALIGNANCY | |||||||
| Malignancy (excluding NMSC) | 0 | 2.2 (2/91.8) | 0 | 1.1 (3/281.8) | |||
| Lymphoma | 0 | 1.1 (1/91.5)a | 0 | 0.4 (1/281.5)a | |||
| NMSC | 0 | 1.1 (1/91.8) | 0.4 (1/268.4) | 0.4 (1/281.8) | |||
| CARDIOVASCULAR EVENTS | |||||||
| Adjudicated VTEb | 0 | 1.1 (1/91.9) | 0.4 (1/268.6) | 0.4 (1/281.9) | |||
| Adjudicated MACEc | 0 | 1.1 (1/91.5) | 0.4 (1/268.6) | 0.4 (1/281.5) | |||
| GASTROENTEROLOGICAL EVENTS | |||||||
| Adjudicated GI perforations | 0 | 0 | 0 | 0 | |||
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
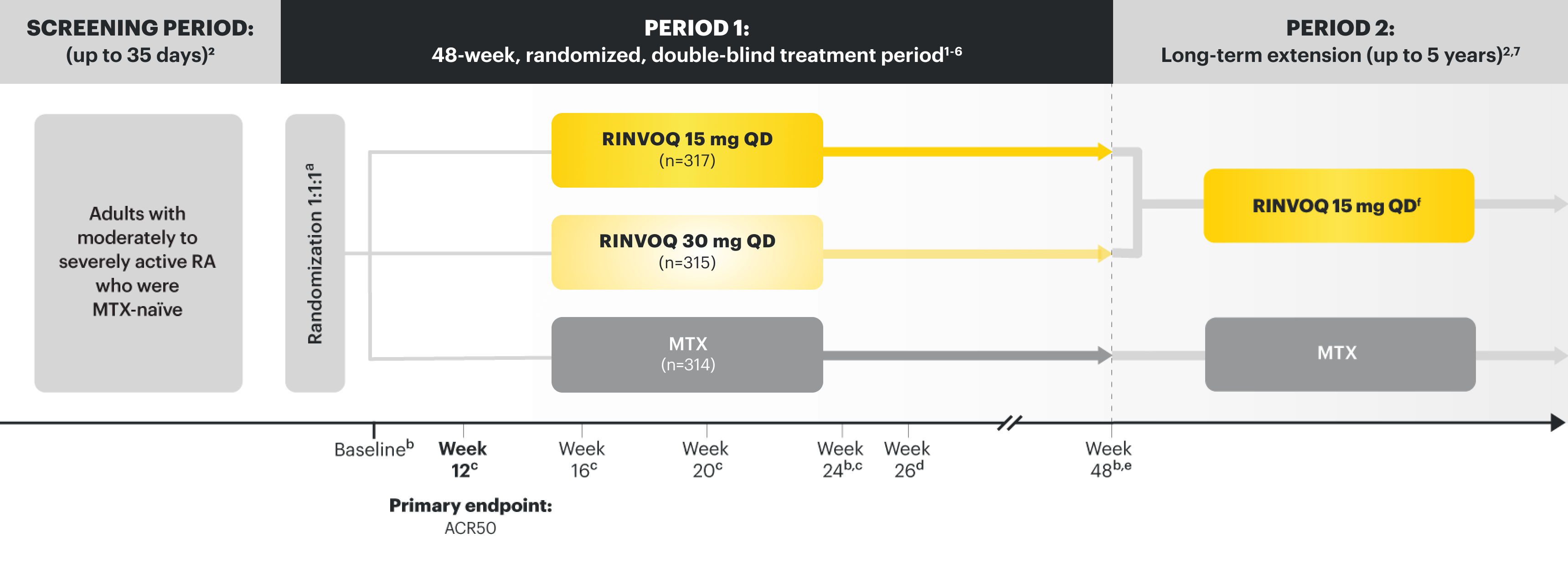
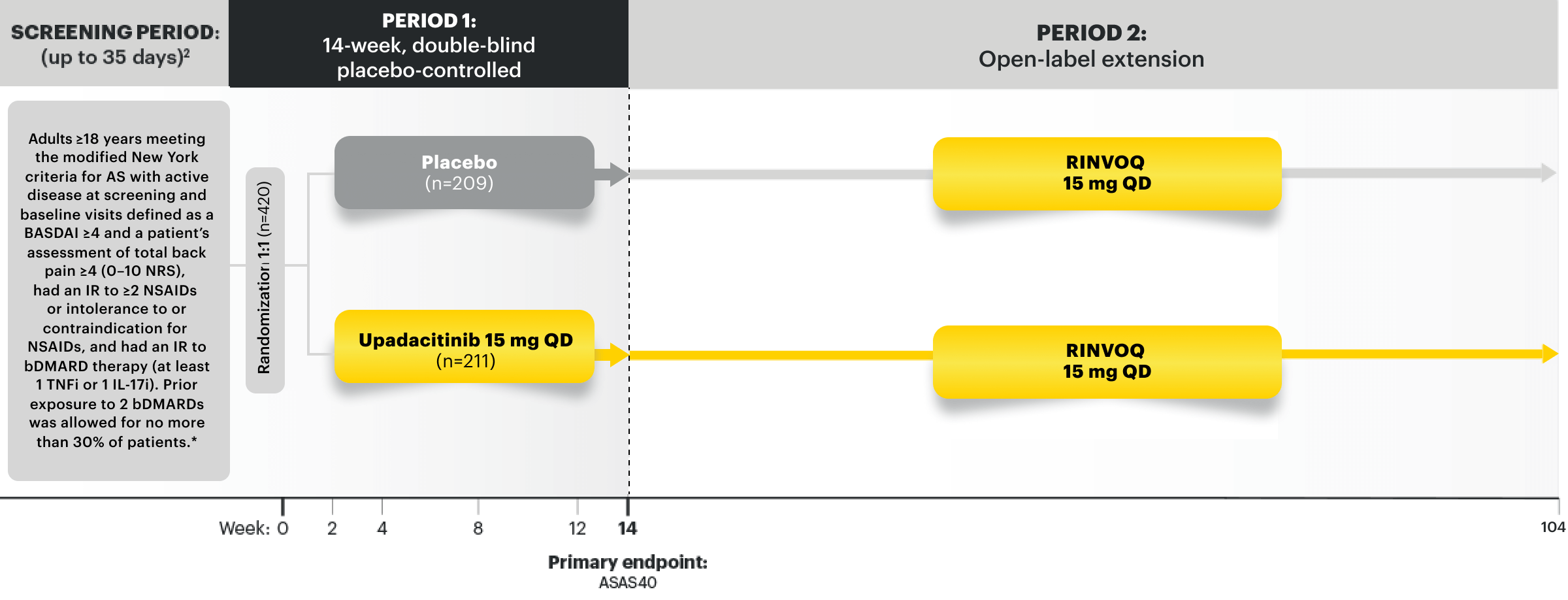
Adverse Events of Special Interest at Week 14 and Week 52 in AS17-20
<<Swipe table to see more
| Week 14 | Week 52 | ||||||
|---|---|---|---|---|---|---|---|
| SELECT-AXIS 2 Study 1 (Phase 3; bDMARD-IR) | SELECT-AXIS 1 (Phase 2/3; bDMARD-naïve) | SELECT-AXIS 2 Study 1 (Phase 3; bDMARD-IR) | |||||
| TEAEs of Special Interest* | Placebo n=209 n (%) |
RINVOQ 15 mg QD n=211 n (%) | Placebo n=94 n (%) |
RINVOQ 15 mg QD n=93 n (%) | Any RINVOQ 15 mg QD n=414 PYs=534.4 E (E/100 PYs) | ||
| INFECTIONS | |||||||
| Serious infections | 0 (0.0) | 5 (2)a | 0 (0.0) | 0 (0.0) | 24 (4.5) | ||
| Active TB | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
| Opportunistic infection (excluding TB and herpes zoster) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1)b | 0 (0.0) | ||
| Herpes zoster | 0 (0.0) | 2 (1)c | 0 (0.0) | 0 (0.0) | 19 (3.6) | ||
| MALIGNANCY | |||||||
| Malignancy (excluding NMSC) | 1 (1)d | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | ||
| Lymphoma | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | ||
| NMSC | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.4) | ||
| CARDIOVASCULAR EVENTS | |||||||
| Adjudicated VTEe | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.4) | ||
| Adjudicated MACEf | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | ||
| GASTROENTEROLOGICAL EVENTS | |||||||
| Adjudicated GI perforations | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||
| Inflammatory bowel diseaseg | 0 (0.0) | 1 (0.5)h | 1 (1.1) | 0(0.0) | 1 (0.2) | ||
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
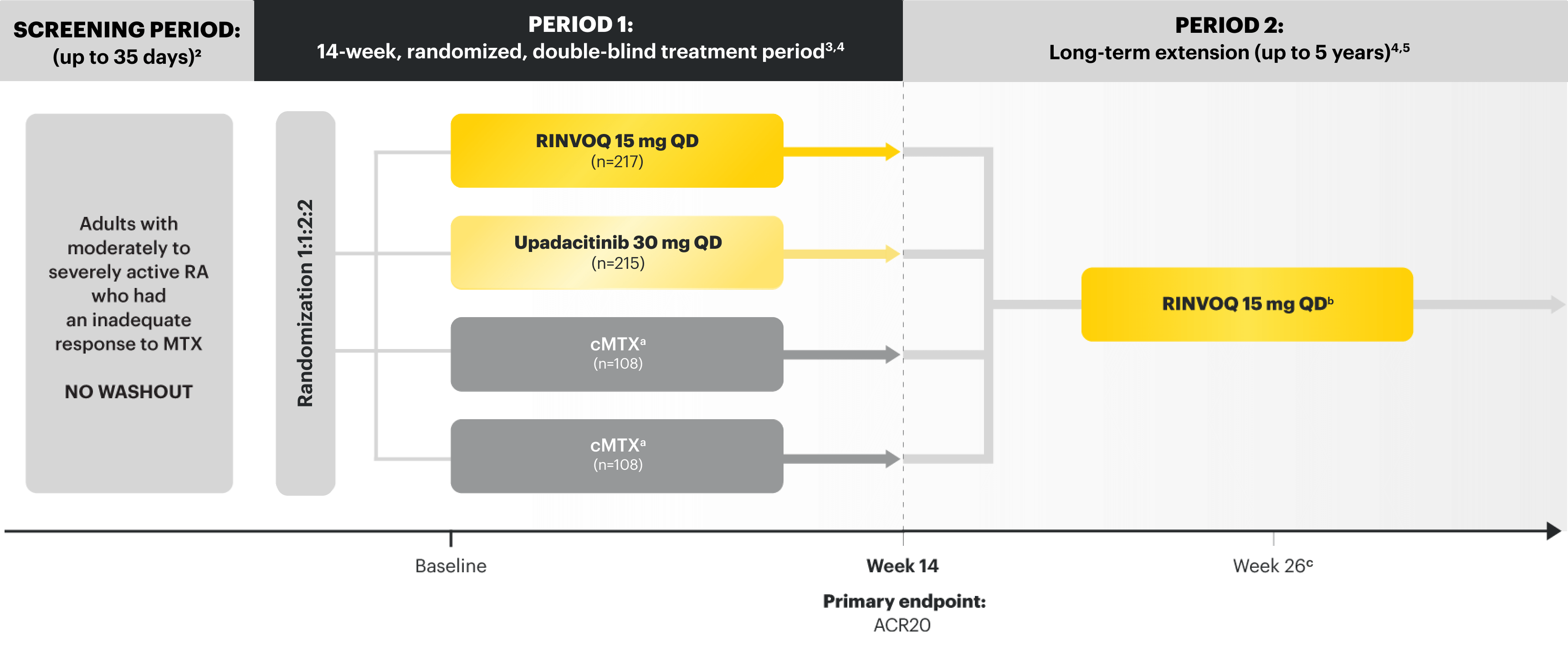
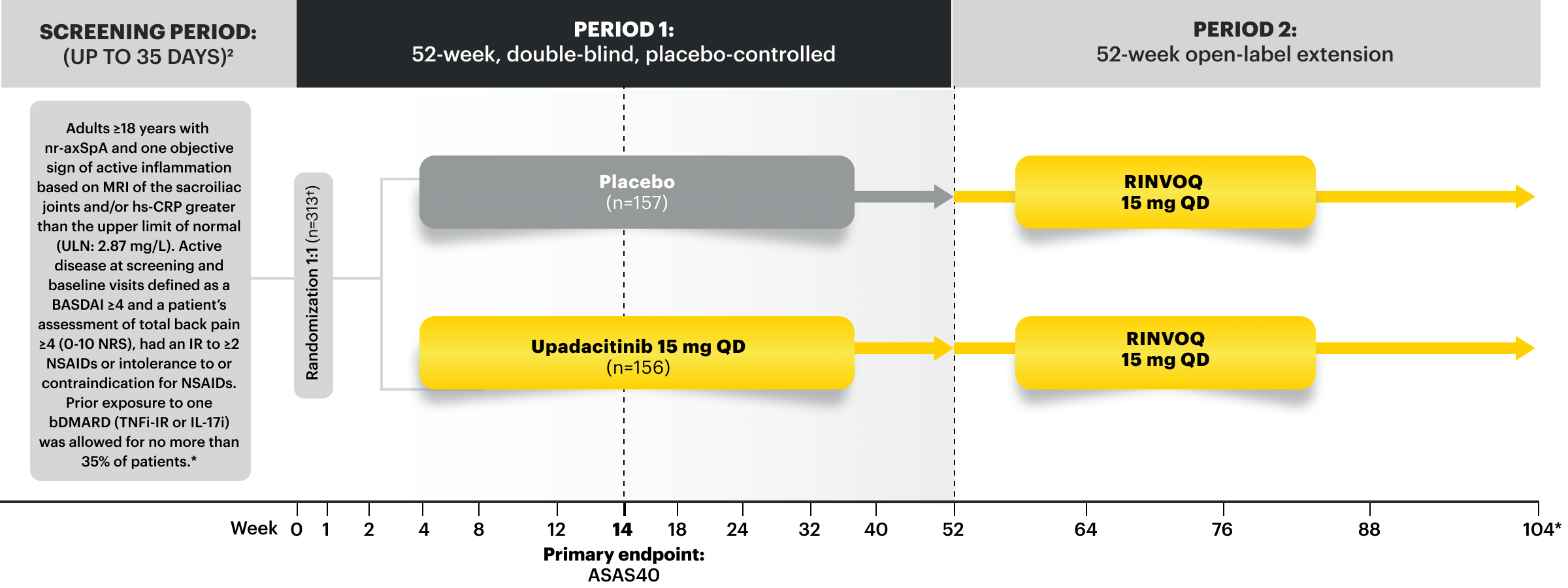
Adverse Events of Special Interest at Week 14 and Week 52 in nr-axSpA22,23
<<Swipe table to see more
| Week 14 | Week 52 | ||||||
|---|---|---|---|---|---|---|---|
| SELECT-AXIS 2 Study 2 (Phase 3; mixed*) | SELECT-AXIS 2 Study 2 (Phase 3; mixed*) | ||||||
| TEAEs of Special Interest† | Placebo n=157 n (%) |
RINVOQ 15 mg QD n=156 n (%) |
Placebo n=157 PYs=140.2 E (E/100 PYs) |
RINVOQ 15 mg QD n=156 PYs=137.4 E (E/100 PYs) |
|||
| INFECTIONS | |||||||
| Serious infections | 1 (1)a | 2 (1)b | 1 (0.7) | 2 (1.5) | |||
| Active (TB) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Opportunistic infection (excluding TB and herpes zoster) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Herpes zoster | 1 (1) | 2 (1)c | 1 (0.7) | 5 (3.6) | |||
| MALIGNANCY | |||||||
| Malignancy (excluding NMSC) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Lymphoma | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.7) | |||
| NMSC | 1 (1)d | 0 (0.0) | 1 (0.7) | 0 (0.0) | |||
| CARDIOVASCULAR EVENTS | |||||||
| Adjudicated VTEe | 0 (0.0) | 0 (0.0) | 2 (1.4) | 0 (0.0) | |||
| Adjudicated MACEf | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| GASTROINTESTINAL EVENTS | |||||||
| Adjudicated GI perforations | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
| Inflammatory bowel diseaseg | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
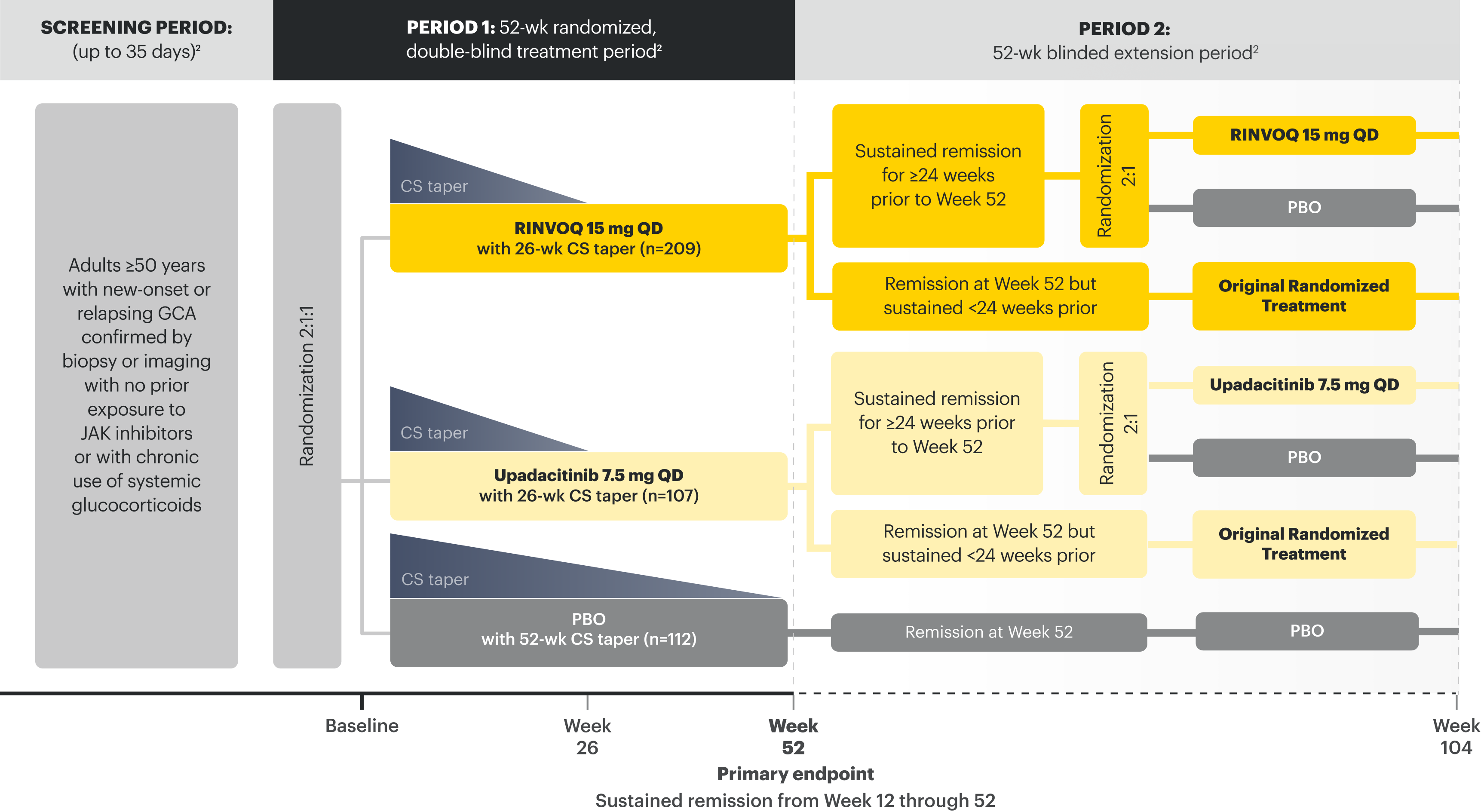
Adverse Events of Special Interest at Week 52 in GCA5,27
| TEAEs of Special Interest* (E/100 PYs unless otherwise stated) | Placebo + 52-wk CS Taper N=112, PYs=94.3 |
RINVOQ 15 mg QD + 26-wk CS Taper N=209, PYs=178.1 |
|---|---|---|
| INFECTIONS | ||
| Serious infections | 12.7 | 7.9 |
| Active tuberculosis (TB) | 0 | 0 |
| Opportunistic infection (excluding TB, herpes zoster, and oral candidiasis) | 1.1 | 2.2 |
| Herpes zoster | 4.2 | 7.3 |
| MALIGNANCYa | ||
| Malignancy (excluding NMSC) | 2.1 | 2.3 |
| Lymphoma | 0 | 0 |
| NMSC | 2.1 | 2.8 |
| CARDIOVASCULAR EVENTSa | ||
| Adjudicated VTEb | 4.3 | 3.9 |
| Adjudicated MACEc | 2.1 | 0 |
| GASTROENTEROLOGICAL EVENTSa | ||
| Adjudicated GI perforations | 0 | 0 |
Safety Considerations
Consider the Benefits and Risks for the Individual Patient Prior to Initiating Therapy with RINVOQ
Consistent Safety Profile of AEs Observed in Long-Term Analysis
RA Phase 3 Program AEs4,7-9*
Any RINVOQ 15 mg QD†
<<Swipe table to see more
| TEAEs of Special Interest‡ (E/100 PYs unless otherwise stated) | 2.8 yrs. max. exposure(1.4 yrs. median) Data as of November 14, 2018 n=2630, PYs=3446.2 |
~6.5 yrs. max. exposure(~4 yrs. median) Data as of August 15, 2022 n=3209, PYs=10782.7 |
~9.5 yrs. max. exposure(-4.2 yrs. median) n=3209, PYs=12909.0 |
|---|---|---|---|
| INFECTIONS | |||
| Serious infections | 3.5 | 3.6 | 3.5 |
| Active tuberculosis (TB) | 0.1 | <0.1 | <0.1 |
| Opportunistic infection (excluding TB, herpes zoster, and oral candidiasis) | 0.3 | 0.3 | 0.2 |
| Herpes zoster | 3.5 | 3.2 | 3.0 |
| MALIGNANCYa | |||
| Malignancy (excluding NMSC) | 0.8 | 0.7 | 0.7 |
| Lymphoma | <0.1 | <0.1 | <0.1 |
| NMSC | 0.3 | 0.4 | 0.4 |
| CARDIOVASCULAR EVENTSa | |||
| Adjudicated VTEb | 0.6 | 0.4 | 0.4 |
| Adjudicated MACEc | 0.6 | 0.3 | 0.4 |
| GASTROENTEROLOGICAL EVENTSa | |||
| Adjudicated GI perforations | 0.1 | <0.1 | <0.1 |
Adverse reaction rates observed in clinical trials and LTE studies may not predict the rates observed in clinical practice.
ADVERSE REACTIONS: The most common adverse reactions in RA RINVOQ clinical trials (≥1%) were upper respiratory tract infections, nausea, cough, pyrexia. Other adverse reactions reported in less than 1% of patients in the RINVOQ 15 mg group and at a higher rate than in the placebo group through Week 12 included pneumonia, herpes zoster, herpes simplex (includes oral herpes), and oral candidiasis.
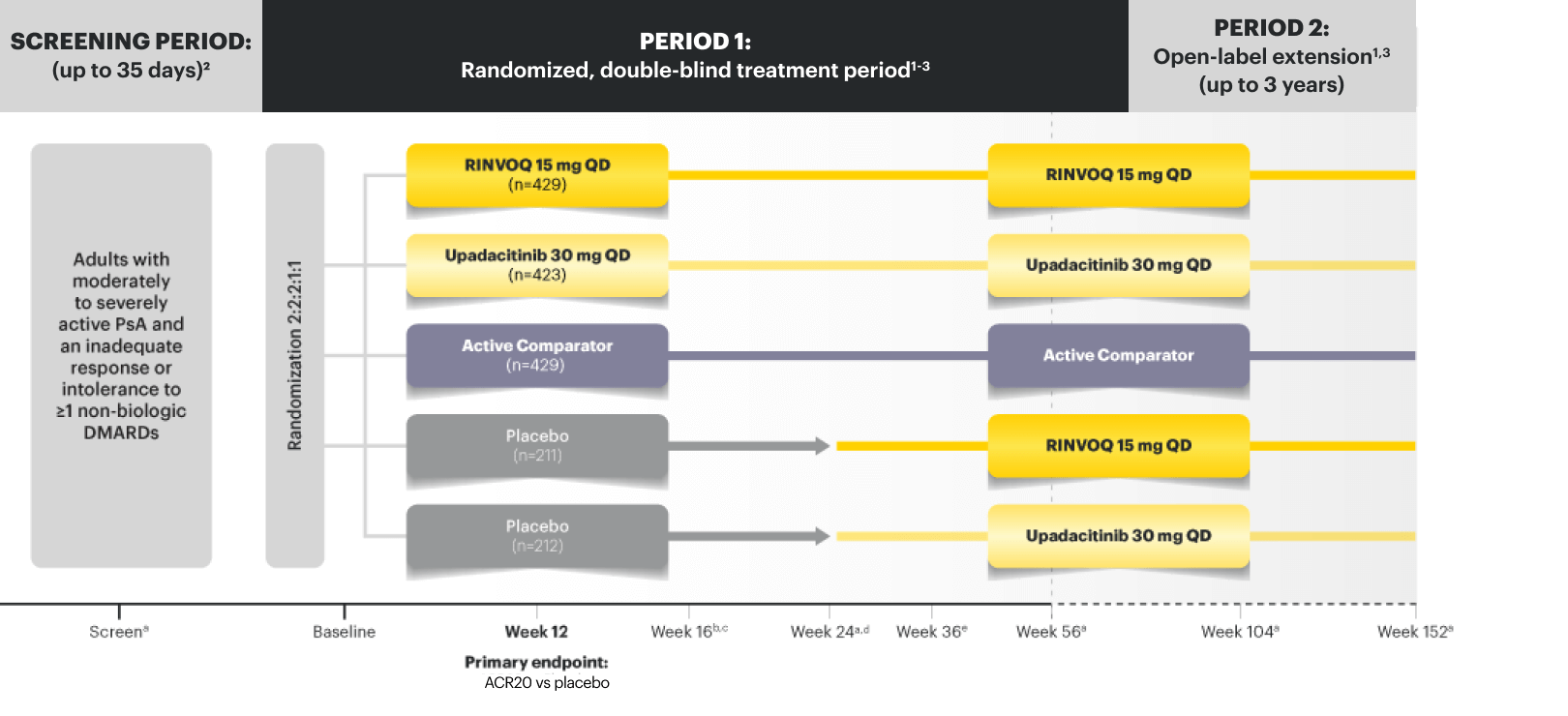
PsA Phase 3 Program AEs4,12-15
Any RINVOQ 15 mg QD*
<<Swipe table to see more
| TEAE of Special Interest† (E/100 PYs unless otherwise stated) | ~3 yrs. max. exposure(-1.3 yrs. median) Data as of June 30, 2020 n=907, PYs=1247.2 |
~4 yrs. max. exposure(-2.3 yrs. median) Data as of June 30, 2021 n=907, PYs=1872.3 |
~6.4 yrs. max. exposure(-3.6 yrs. median) n=907, PYs=2974.9 |
|---|---|---|---|
| INFECTIONS | |||
| Serious infection | 2.3 | 3.9 | 3.3 |
| Active tuberculosis (TB) | 0 | 0 | 0 |
| Opportunistic infection (excluding TB, herpes zoster, and oral candidiasis) | 0.4 | 0.5 | 0.3 |
| Herpes zoster | 3.8 | 3.6 | 3.1 |
| MALIGNANCYa | |||
| Malignancy (excluding NMSC) | 0.7 | 0.6 | 0.8 |
| Lymphomab | 0.2 | 0.1 | 0.1 |
| NMSC | 0.7 | 0.6 | 0.7 |
| CARDIOVASCULAR EVENTSa | |||
| Adjudicated VTEc | 0.3 | 0.2 | 0.2 |
| Adjudicated MACEd | 0.3 | 0.3 | 0.4 |
| GASTROENTEROLOGICAL EVENTSa | |||
| Adjudicated GI perforations | <0.1 | 0.1 | <0.1 |
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
No Warnings or Precautions for IBD‡
with RINVOQ in the FDA-approved label.1§
BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis with additional Warnings and Precautions including Hypersensitivity reactions, Gastrointestinal perforations, Lab abnormalities, Embryo-fetal toxicity, Vaccinations, and Medication residue in stool.
§Does not preclude events from occurring in RINVOQ-treated patients.
AS Phase 2/3 & 3 Program AEs4
Any RINVOQ 15 mg QD
<<Swipe table to see more
| TEAEs of Special Interest* (E/100 PYs unless otherwise stated) | ~3.8 years max. exposure (-1.8 years median) n=596, PYs=1016.6 |
|---|---|
| INFECTIONS | |
| Serious infections | 2.5 |
| Active tuberculosis (TB) | 0 |
| Opportunistic infection (excluding TB, herpes zoster, and oral candidiasis) | 0.2 |
| Herpes zoster | 3.1 |
| MALIGNANCYa | |
| Malignancy (excluding NMSC) | 0.2 |
| Lymphomab | <0.1 |
| NMSC | 0.3 |
| CARDIOVASCULAR EVENTSa | |
| Adjudicated VTEc | 0.4 |
| Adjudicated MACEd | 0.2 |
| GASTROENTEROLOGICAL EVENTSa | |
| Adjudicated GI perforations | 0 |
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
No Warnings or Precautions for IBD†
with RINVOQ in the FDA-approved label.1‡
BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis with additional Warnings and Precautions including Hypersensitivity reactions, Gastrointestinal perforations, Lab abnormalities, Embryo-fetal toxicity, Vaccinations, and Medication residue in stool.
‡Does not preclude events from occurring in RINVOQ-treated patients.
nr-axSpA Phase 3 Program AEs4
| TEAEs of Special Interest* (E/100 PYs unless otherwise stated) | RINVOQ 15 mg QD ~2.3 years max. exposure (~1.0 year median) n=286, PYs=380.9 |
|---|---|
| INFECTIONS | |
| Serious infections | 1.3 |
| Active tuberculosis (TB) | 0 |
| Opportunistic infection (excluding TB, herpes zoster, and oral candidiasis) | 0.3 |
| Herpes zoster | 2.6 |
| MALIGNANCYa | |
| Malignancy (excluding NMSC) | 0.3 |
| Lymphoma | 0.3 |
| NMSC | 0.3 |
| CARDIOVASCULAR EVENTSa | |
| Adjudicated VTEb | 0.8 |
| Adjudicated MACEc | 0.5 |
| GASTROENTEROLOGICAL EVENTSa | |
| Adjudicated GI perforations | 0 |
Adverse reaction rates observed in clinical trials may not fully characterize the risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
No Warnings or Precautions for IBD†
with RINVOQ in the FDA-approved label.1‡
BOXED WARNING on Serious Infections, Mortality, Malignancies, Major Adverse Cardiovascular Events, and Thrombosis with additional Warnings and Precautions including Hypersensitivity reactions, Gastrointestinal perforations, Lab abnormalities, Embryo-fetal toxicity, Vaccinations, and Medication residue in stool.
‡Does not preclude events from occurring in RINVOQ-treated patients.
GCA Phase 3 Program AEs5,27-29
<<Swipe table to see more
| Week 52 | Week 104 | ||||||
|---|---|---|---|---|---|---|---|
| ~1.0 years max. exposure (~1.0 years median) |
~2.0 years max. exposure (~2.0 years median) |
||||||
| TEAE of Special Interest* (E/100 Pys unless otherwise stated) |
Placebo + 52‑wk CS Taper N=112, PYs=94.3 |
RINVOQ 15 mg QD + 26‑wk CS Taper N=209, PYs=178.1 |
Placebo + 52‑wk CS Taper N=112, PYs=133.9 |
RINVOQ 15 mg QD + 26‑wk CS Taper N=209, PYs=270.3 |
|||
| INFECTIONS | |||||||
| Serious Infections | 12.7 | 7.9 | 10.5 | 5.9 | |||
| Active Tuberculosis (TB) | 0 | 0 | 0 | 0 | |||
| Opportunistic Infection (excluding TB and Herpes Zoster) | 1.1 | 2.2 | 1.5 | 1.8 | |||
| Herpes Zoster | 4.2 | 7.3 | 3.0 | 5.9 | |||
| MALIGNANCYa | |||||||
| Malignancy (excluding NMSC) | 2.1 | 2.3 | 1.5 | 1.9 | |||
| Lymphoma | 0 | 0 | 0 | 0 | |||
| NMSC | 2.1 | 2.8 | 3.8 | 2.6 | |||
| CARDIOVASCULAR EVENTSa | |||||||
| Adjudicated VTEb | 4.3 | 3.9 | 3.0 | 3.0 | |||
| Adjudicated MACEc | 2.1 | 0 | 1.5 | 0 | |||
| GASTROENTEROLOGICAL EVENTSa | |||||||
| Adjudicated GI Perforations | 0 | 0 | 0 | 0 | |||
Adverse reaction rates observed in clinical trials may not fully characterize that risks of RINVOQ. Certain adverse events may require longer observation periods and longer-term patient exposure to ascertain risk.
In the SELECT-GCA Trial, No New Safety Signals* were observed, including no events of MACE or GI perforations** with RINVOQ 15 mg through Week 104.
In SELECT-GCA at baseline:
- Mean age was 71
- 84% had at least 1 CV risk factor
- 46% were current or former smokers
BOXED WARNING (MACE): Consider the benefits and risks for a patient prior to initiating or continuing therapy with RINVOQ, particularly in patients who are current or past smokers and those with other CV risk factors. Warning and Precaution (GI perforations): Gastrointestinal perforations have been reported in clinical trials with RINVOQ.
Please see below for additional information about these warnings.
*A signal is an observed association between a product and an event. A definitive RINVOQ safety outcomes trial has not been conducted.
**Does not preclude events from occurring in RINVOQ-treated patients.
Well-Studied Safety Profile
Most Common Adverse Reactions From RINVOQ Clinical Trials
RHEUMATOID ARTHRITIS1
Most Common Adverse Reactions From SELECT-BEYOND, SELECT-NEXT, and SELECT-COMPARE (12 Weeks)1
Adverse reactions reported in ≥1% of moderate to severe rheumatoid arthritis patients treated with RINVOQ 15 mg pooled from the placebo‑controlled studies.
| ADVERSE REACTION | Placeboa n=1042 (%) | RINVOQ 15 mg QDa n=1035 (%) |
|---|---|---|
| Upper respiratory tract infection (URTI)b | 9.5 | 13.5 |
| Nausea | 2.2 | 3.5 |
| Cough | 1.0 | 2.2 |
| Pyrexia | 0 | 1.2 |
INFECTIONS
- In the placebo‑controlled studies SELECT-BEYOND, SELECT-NEXT, and SELECT-COMPARE through 12/14 weeks, infections were reported in 20.9% of patients treated with placebo and 27.4% in patients treated with RINVOQ 15 mg10
- In the 12-month exposure dataset, the incident rate of infection was 83.8 per 100 patient-years for patients treated with RINVOQ 15 mg10
ADVERSE REACTIONS: The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenza-like illness, elevated liver enzymes, and rash.1
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.1
PSORIATIC ARTHRITIS11
Most Common Adverse Reactions From SELECT‑PsA 1 and SELECT‑PsA 2 (24 Weeks)
Adverse reactions reported in >1% of psoriatic arthritis patients treated with RINVOQ 15 mg pooled from the placebo‑controlled studies.
| ADVERSE REACTION | Placeboa n=635 (%) | RINVOQ 15 mg QDa n=640 (%) |
|---|---|---|
| Upper respiratory tract infection (URTI)b | 6.9 | 8.9 |
| Bronchitis | 2.2 | 3.8 |
| Cough | 1.6 | 2.5 |
| Nausea | 2.4 | 2.0 |
| Herpes simplexc | 1.2 | 1.4 |
| Acne | 0.3 | 1.3 |
| Herpes zoster | 0.8 | 1.1 |
INFECTIONS
- In the placebo-controlled studies SELECT-PsA 1 and SELECT-PsA 2 through 24 weeks, infections were reported in 33.5% of patients treated with placebo and 37.5% of patients treated with RINVOQ 15 mg16
ADVERSE REACTIONS: The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenza-like illness, elevated liver enzymes, and rash.1
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.1
ANKYLOSING SPONDYLITIS21
Most Common Adverse Reactions From SELECT-AXIS 2 Study 1 (14 Weeks)
Adverse reactions reported in >2% of ankylosing spondylitis patients treated with RINVOQ 15 mg from the placebo-controlled study.21
| ADVERSE REACTION | Placeboa n=209 (%) | RINVOQ 15 mg QDa n=211 (%) |
|---|---|---|
| COVID-19 | 1.9 | 3.3 |
| Headache | 1.4 | 3.3 |
| Neutropenia | 0.5 | 2.8 |
| Hyperuricemia | 0 | 2.4 |
| Nasopharyngitis | 1.4 | 2.4 |
INFECTIONS
- In the placebo-controlled study SELECT-AXIS 2 Study 1 through 14 weeks, infections were reported in 13% of patients treated with placebo and 15% of patients treated with RINVOQ 15 mg17
ADVERSE REACTIONS: The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenza-like illness, elevated liver enzymes, and rash.1
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.1
NON-RADIOGRAPHIC AXIAL SPONDYLOARTHRITIS24
Most Common Adverse Reactions From SELECT-AXIS 2 Study 2 (14 Weeks)
Adverse reactions reported in >2% of non-radiographic axial spondyloarthritis patients treated with RINVOQ 15 mg from the placebo-controlled study.24
| ADVERSE REACTION | Placeboa n=157 (%) | RINVOQ 15 mg QDa n=156 (%) |
|---|---|---|
| Headache | 2.5 | 5.8 |
| Nausea | 1.9 | 3.2 |
| Abdominal pain | 0.6 | 2.6 |
| Diarrhea | 1.9 | 2.6 |
| Neutropenia | 0 | 2.6 |
anr-axSpA patients could be on background NSAIDs.
INFECTIONS
- In the placebo-controlled study SELECT-AXIS 2 Study 2 through 14 weeks, infections were reported in 23% of patients treated with placebo and 23% of patients treated with RINVOQ 15 mg22
ADVERSE REACTIONS: The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenza-like illness, elevated liver enzymes, and rash.1
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.1
GIANT CELL ARTERITIS1
Most Common Adverse Reactions from SELECT-GCA (52 Weeks)
Adverse reactions reported in ≥5% of GCA patients treated with RINVOQ 15 mg + 26-wk CS taper.
| ADVERSE REACTION | Placebo + 52-wk CS Taper n=112 (%) | RINVOQ 15 mg QD + 26-wk CS Taper n=209 (%) |
|---|---|---|
| Upper respiratory tract infection (URTI)a | 20.5 | 21.5 |
| Headache | 11.6 | 16.3 |
| Fatigue | 5.4 | 9.1 |
| Peripheral edemab | 2.7 | 8.6 |
| Coughc | 3.6 | 7.2 |
| Anemiad | 2.7 | 6.7 |
| Rashe | 2.7 | 5.7 |
| Herpes zosterf | 2.7 | 5.3 |
| Nausea | 3.6 | 5.3 |
INFECTIONS
- In the placebo-controlled study SELECT-GCA through 52 weeks, infections were reported in 58.9% of patients treated with placebo + 52-wk CS taper and 63.2% of patients treated with RINVOQ 15 mg + 26-wk CS taper.5
ADVERSE REACTIONS: The most common adverse reactions in RINVOQ clinical trials were upper respiratory tract infections, herpes zoster, herpes simplex, bronchitis, nausea, cough, pyrexia, acne, headache, peripheral edema, increased blood creatine phosphokinase, hypersensitivity, folliculitis, abdominal pain, increased weight, influenza, fatigue, neutropenia, myalgia, influenza-like illness, elevated liver enzymes, rash, and anemia.1
Inform patients that retinal detachment has been reported in clinical trials with RINVOQ. Advise patients to immediately inform their healthcare provider if they develop any sudden changes in vision while receiving RINVOQ.1